У болези Паркинсона и старения общие причины
У болези Паркинсона и старения общие причины
http://elementy.ru/news/430260
|
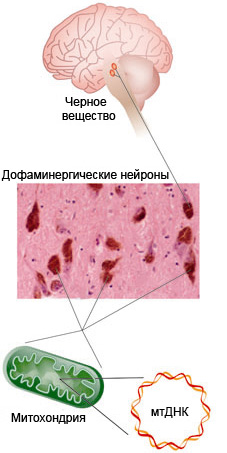
Недавно две группы ученых убедительно доказали, что одной из причин старения и болезни Паркинсона являются дефектные митохондрии, быстро размножающиеся в нейронах головного мозга и приводящие к их дегенерации. Дефектными они становятся в результате соматической мутации — удаления участка митохондриальной ДНК. Однако остается неясным, почему с возрастом дефектные митохондрии появляются в определенных нейронах головного мозга.
Пока нет общепринятого мнения о причинах старения. По одной из гипотез, старение и смерть индивидуумов, не способных более к размножению, выгодно для вида, поскольку экономятся ресурсы (пища, территория), необходимые для молодых, более полезных для существования всего вида особей. Хотя не известно точно, сколько генов задействовано в этой программе старения и как они взаимодействуют, но, детально изучив их, можно попытаться заблокировать механизм старения.
Согласно же другой гипотезе, старение — результат действия сотен или даже тысяч вредных генетических признаков, которые начинают проявляться лишь по окончании репродуктивного периода. Мы будем называть их DILL-признаки (DILL — от англ. Deleterious In Late Life), или DILL-аллели.
Почему эти DILL-признаки накапливались в течение эволюции? Выделяют три возможные причины (Cortopassi 2002). Во-первых, численность популяции на протяжении большей части эволюции человека была крайне низкой (примерно 10 000), в связи с чем случайные процессы сильно влияли на эволюцию генов человека и приводили к закреплению множества вредных аллелей. Недавнее резкое увеличение численности популяции еще не отразилось на эволюции генов человека.
Во-вторых, мутация, проявляющаяся лишь после репродуктивного периода, очень слабо влияет на приспособленность организма (число детей) и поэтому может распространиться в популяции случайным образом, без влияния естественного отбора.
И в-третьих, даже если предположить, что DILL-мутации как-то
влияют на приспособленность (например, мудрые советы пожилых людей
обеспечивают лучшую выживаемость их детей и внуков), естественный
отбор не мог элиминировать эти мутации, поскольку в древней
человеческой популяции средняя продолжительность жизни была низкой.
За 5 миллионов лет могло накопиться множество мутаций,
вызывающих ухудшение слуха в 50 лет, ослабление памяти
в 55, болезнь Паркинсона в 60, дряблость кожи в 65...
Все они находились в полной безвестности, пока примерно
10 000 лет назад человек не занялся земледелием,
продолжительность его жизни увеличилась, и все эти DILL-мутации
стали проявляться. Начиная с этого момента человек стал умирать
преимущественно от внутренних причин
Однако гены накопили DILL-мутации неравномерно, поэтому и потенциальная вредность от этих генов в старости будет разная. Например, митохондриальные гены (те которые содержатся в митохондрии, наследуемой через цитоплазму яйцеклетки от матери) должны содержать много DILL-мутаций. Это объясняется тем, что в связи с гаплоидностью, цитоплазматическим наследованием и отсутствием рекомбинации митохондриальные гены обладают низкой эффективной численностью, поэтому на их эволюцию сильно влияют случайные факторы и слабо влияет естественный отбор. В результате дрейфа генов в малочисленной популяции митохондриальных генов могут фиксироваться неоптимальные аллели, содержащие вредные мутации.
Кроме того, митохондрии обладают повышенным темпом мутирования, что увеличивает вероятность появления новой соматической мутации в течение жизни человека. Но, поскольку в каждой клетке содержатся тысячи митохондриальных геномов (в среднем около 2000), они создают генетическую избыточность, и один появившийся мутант, сосуществуя с нормальными митохондриальными геномами, не может сильно повлиять на приспособленность клетки. Дальнейшая судьба мутантного генома зависит от соотношения сил на внутриклеточном и межклеточном уровнях (Taylor 2002).
Внутри клетки митохондриальные геномы конкурируют друг с другом. Геномы, которые быстрее реплицируются, увеличивают свою численность внутри клетки и вытесняют медленно-размножающихся конкурентов. Если у одного генома произошла крупная делеция (вырезание участка ДНК), то он становится нефункциональным (так как производится меньше АТФ — энергетической молекулы, необходимой для ускорения большинства биологических реакций), но зато коротким и быстро размножающимся — и такой эгоистичный геном победит во внутриклеточной борьбе.
Однако межклеточный уровень конкуренции ставит всё на свои места: клетка, в которой мутантные митохондриальные геномы достигли высокой концентрации, менее жизнеспособна и медленнее делится по сравнению с клеткой с нормальными митохондриальными геномами, и мутантные геномы элиминируются из организма вместе со своей клеткой.
Важно отметить, что порог проявления патогенного эффекта мутантного генома варьирует в зависимости от энергетической потребности клетки. Так, ткани, сильно зависящие от митохондриального метаболизма (мышцы, сердце и нервная система) более чувствительны к накоплению мутантных геномов и, значит, имеют самый низкий порог.
Помимо своей высокой энергетической емкости нервная ткань характеризуется чрезвычайно медленным делением клеток, что приводит к тому, что межклеточный уровень отбора оказывается слабее внутриклеточного и с большой вероятностью может проявиться патогенный эффект, вызванный мутантными митохондриальными геномами.
Исследуя мутации в митохондриальной ДНК (мтДНК) нейронов мозга, две независимых лаборатории (Bender et al. 2006; Kraytsberg et al. 2006) получили интересные результаты о причинах старения и болезни Паркинсона — одной из основных нейродегенеративных болезней старости, характеризующейся нарушением планирования действий, что выражается в акинезии (ограничение произвольных движений), ригидности (повышение мышечного тонуса) и тремор конечностей (дрожание). Результаты их работы опубликованы в майском номере Nature Genetics.
Объектом исследований был головной мозг умерших людей, а именно участок под названием черное вещество (substantia nigra). Черное вещество находится в базальных ганглиях головного мозга (под корой больших полушарий), основная функция которых — регуляция произвольных движений: перехода от замысла (подготовки действия) к выполнению выбранной программы действия. При паркинсонизме происходит дегенерация нейронов черного вещества, выделяющих дофамин и регулирующих таким образом работу базальных ганглиев в целом. Конкретная причина дегенерации дофаминергических нейронов до сих пор была неизвестна.
|
Техническим ноу-хау, позволившим сделать новые открытия, оказалась возможность исследования мутаций в митохондриальной ДНК (мтДНК) и доли мутантных геномов в каждом конкретном нейроне черного вещества, а не во всей нервной ткани.
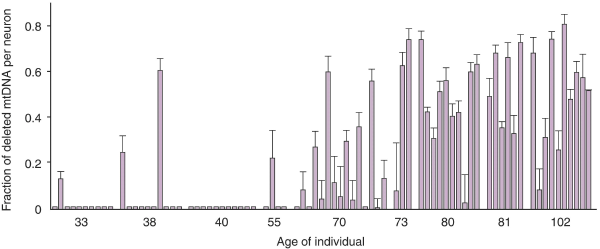
Исследования нейронов черного вещества показали быстрое
накопление делеций с возрастом. Оказалось также, что некоторые
нейроны имеют сильный дефицит
|
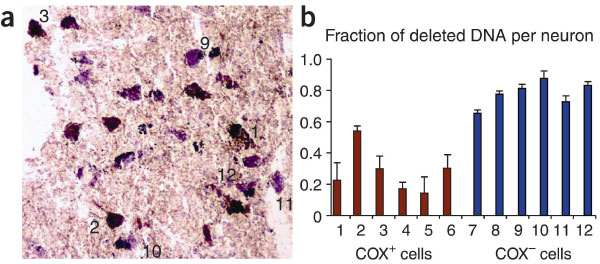
Важно отметить, что разные нейроны содержат уникальные делеции, что говорит о соматическом происхождении этих мутаций в нейронах. Интересно, что нейроны, выделенные из соседней области головного мозга (гиппокамп) старых индивидуумов не показывали высокого уровня делеций. Возможно, повышенная фоновая активность нейронов черного вещества сопряжена с образованием активных форм кислорода, которые, являясь мутагенами, приводят к образованию делеций в мтДНК. Активные формы кислорода могут появляться также и при метаболизме дофамина. Основываясь на этом предположении, для предотвращения появления делеций врачи предлагают обрабатывать нейроны черного вещества антиоксидантными препаратами.
Итак, соматические делеции в мтДНК играют важную роль в
деградации нейронов при старении и болезни Паркинсона. Поскольку
делеции появляются независимо в разных нейронах черного вещества
примерно в одно и тоже время и затрагивают примерно одно и то же
место, скорее всего существуют определенные
Другая потенциальная DILL-мутация может находиться в митохондрильной ДНК-полимеразе — ферменте, который реплицирует мтДНК. Мутантная форма данного фермента приводит к большому числу ошибок при репликации мтДНК, и линии мышей с таким ферментом страдают от раннего старения из-за накопления большого числа мутаций в мтДНК (Trifunovic et al. 2004).
Скорее всего, болезнь Паркинсона, как и старение, может быть вызвана многочисленными причинами. Например, мутация в ядерно-кодируемом гене альфа-синуклеине приводит к слипанию мутантных белков и образованию цитоплазматических включений, характерных для болезни Паркинсона (Polymeropoulos et al. 1997). Сходный эффект проявляется при нарушении у мышей автофагии — процесса удаления старых белков из цитоплазмы клеток (Hara et al. 2006).
Успех в изучении и лечении каждой из причин болезни Паркинсона и старения вносит свой вклад в общее дело борьбы с этим недугом. Впервые болезнь Паркинсона была описана в 1817 году Джеймсом Паркинсоном. Через век — в 1919 году — была предложена заместительная терапия посредством введения в кровь предшественника дофамина, что компенсирует функцию дегенерировавших нейронов черного вещества. В наше время научно доказана причина дегенерации нейронов и обсуждается возможность предотвращения появления этих делеций посредством антиоксидантов (Bender et al. 2006; Kraytsberg et al. 2006). Хочется верить, что к концу XXI века люди смогут приблизиться к причине еще ближе и научатся удалять DILL-мутации непосредственно из генома.
Источники:
1) Bender et al. High levels of mitochondrial DNA deletions in
substantia nigra neurons in aging and Parkinson disease //
Nature genetics. 2006. V. 38. P. 515-517.
2)
Kraytsberg et al. Mitochondrial DNA deletions are abundant and cause
functional impairment in aged human substantia nigra
neurons // Nature genetics. 2006. V. 38.
P. 518-520.
3) G. Manfredi. mtDNA clock runs out for dopaminergic
neurons // Nature genetics. 2006. V. 38.
P. 507-508.